Gjenet që janë pjesë e 22 çifteve të kromozomeve jo seksuale quhen autosomale. Sëmundjet autosomale dominante janë sëmundje në të cilat një gjen mutant (alel) në gjendje heterozigote mjafton për shfaqjen e manifestimeve fenotipike.
Trashëgimia autosomale dominante e sëmundjeve karakterizohet nga një sërë shenjash të mëposhtme, të cilat zbulohen në shumicën e rasteve:
- sëmundja transmetohet vertikalisht përgjatë origjinës, dhe rastet e sëmundjes diagnostikohen në çdo brez;
- rreziku për të trashëguar sëmundjen për cilindo nga fëmijët e pacientit është 50%;
- anëtarët fenotipikë normalë të familjes nuk trashëgojnë sëmundje tek pasardhësit e tyre;
- të dy gjinitë preken me frekuencë të barabartë;
- një pjesë e konsiderueshme e rasteve të sëmundjes shkaktohen nga një mutacion i ri.
Me trashëgiminë autosomale dominante, ndryshe nga tipi i lidhur me X, transmetimi i sëmundjes përmes linjës mashkullore është i mundur. Meqenëse një burrë kalon një kromozom Y dhe jo një kromozom X tek djemtë e tij, në rastet kur një sëmundje trashëgimore kalon nga babai te djali, trashëgimia e lidhur me X është e përjashtuar.
Me trashëgiminë autosomale dominante, ka ndryshueshmëri të konsiderueshme në manifestimet klinike brenda familjes. Në shumicën e rasteve, kjo është për shkak të shprehjes së ndryshueshme të gjenit mutant. Shkaku i saktë i kësaj ndryshueshmërie është i panjohur, por ka shumë të ngjarë që të jetë për shkak të ndikimit të gjeneve modifikuese dhe faktorëve mjedisorë në fenotip. Në disa familje, bartësit e detyrueshëm të gjenit mutant nuk kanë manifestime fenotipike të sëmundjes. Ky fenomen i trashëgimisë autosomale dominante quhet depërtim jo i plotë, d.m.th. trashëgimia ndjek parimin "gjithçka ose asgjë". Në disa raste, kur ka përshtypjen e mungesës së penetrencës, pacienti mund të ketë një shkallë të ulët të mozaicizmit somatik ose mozaicizmit të qelizave germinale për këtë gjen. Mozaicizmi somatik ndodh në fazën e zhvillimit embrional për shkak të një mutacioni në një qelizë somatike, duke çuar në formimin e gjenotipeve të përziera në qelizat fetale, disa prej të cilave e përmbajnë mutacionin, ndërsa të tjerëve u mungon. Në rastet tipike, efektet e gjenit mutant janë më pak të theksuara ose mungojnë në këta pacientë. Mozaicizmi i qelizave germinale ndodh në embrion pas konceptimit dhe kufizohet në qelizat që janë pararendëse të vezëve ose spermës. Shpesh vërehet në kushte të tilla si osteogenesis imperfecta dhe sindroma të shoqëruara me kraniostenozë (sindromat Apert dhe Crouzon).
Meqenëse një gjen i vetëm mutant është i mjaftueshëm për manifestimet fenotipike të trashëgimisë autosomale dominante, këto kushte lindin në shumë pacientë si rezultat i një mutacioni të ri. Sa më e rëndë të jetë sëmundja, aq më e madhe është incidenca e rasteve që vijnë nga mutacionet e gjeneve de novo. Në sëmundjet e rënda, ulja e funksionit riprodhues kufizon transmetimin e gjenit mutant. Në disa raste të shfaqjes së një mutacioni të ri, prindërit janë të moshuar (mbi 40 vjeç).
Artikulli është përgatitur dhe redaktuar nga: kirurgVideo:
Të shëndetshëm:
Artikuj të ngjashëm:
- Teorikisht, individët që mbartin një tipar dominues mund të jenë homozigotë ose heterozigotë. Megjithatë, kur bëhet fjalë për...
- Sëmundjet autosomale recesive përfshijnë sëmundjet në të cilat duhen dy kopje të një mutanti për shfaqjen e karakteristikave fenotipike...
Sëmundjet më tipike me një lloj trashëgimie autosomale recesive janë fibroza cistike, fenilketonuria, galaktosemia, sindroma adrenogjenitale dhe mukopolisakaridoza. Karakteristikat e sëmundjeve të lidhura me seksin janë për shkak të faktit se gratë kanë dy kromozome X dhe burrat kanë një. Një grua merr dy kromozomet e saj X dhe gjenet përkatëse nga babai dhe nëna e saj, ndërsa një burrë trashëgon kromozomin e vetëm X vetëm nga nëna e tij. Një grua, pasi ka trashëguar një gjen patologjik nga njëri prej prindërve të saj, është heterozigot, dhe një burrë është hemizigot, pasi gjenet e vendosura në kromozomin X nuk kanë alele në kromozomin Y. Në këtë drejtim, tiparet e trashëguara në një mënyrë të lidhur me X ndodhin në një popullatë me probabilitete të ndryshme në meshkuj dhe femra. Trashëgimia e lidhur me kromozomet seksuale mund të jetë dominuese ose recesive (zakonisht recesive).
Lloji i trashëgimisë është autosomik dominant. llojet e trashëgimisë së tipareve te njerëzit
Disa sëmundje gjenetike dominuese shfaqen menjëherë pas lindjes. Të tjerat mund të mos shfaqen deri në moshën madhore.
Kujdes
Shembuj të sëmundjeve të tilla janë sëmundja policistike e veshkave tek të rriturit dhe korea e Huntington-it. Si trashëgohen sëmundjet dominuese? Figura 2: Si transmetohen sëmundjet mbizotëruese nga prindi tek fëmija Nëse njëri nga prindërit ka një kopje të ndryshuar të gjenit, atëherë ai mund t'ia kalojë fëmijës ose kopjen normale ose kopjen e ndryshuar.
Kështu, secili prej fëmijëve të një prindi të tillë do të kishte një shans 50% për të trashëguar kopjen e ndryshuar dhe për rrjedhojë të ketë sëmundjen gjenetike.
Lloji autosomik dominant i trashëgimisë
Normalisht, gjenet hemizigotë janë ato të lokalizuara në kromozomet seksuale të seksit heterogametik, d.m.th., seksi që prodhon lloje të ndryshme të qelizave germinale. Hemizigoziteti ndodh gjithashtu si rezultat i aneuploidisë ose delecionit, kur vetëm një nga një palë gjenesh alelike ruhet në gjenotip, i cili mund të shfaqet si një mutacion recesiv.
Sëmundjet e karakterizuara nga trashëgimia mbizotëruese e lidhur me X përfshijnë rakitin rezistent ndaj vitaminës D (rakit që nuk mund të trajtohet me doza të rregullta të vitaminës D), sindromën orofacial-dixhitale (frenulum i shumëfishtë hiperplastik i gjuhës, çarje e buzës dhe qiellzës, hipoplazia alar e hundës, asimetrike shkurtimi i gishtave) dhe sëmundje të tjera.
Trashëgimia dominuese
Në një nivel të ulët depërtimi, gjeni mutant mund të mos shfaqet në çdo brez. Më shpesh, lloji i trashëgimisë është autosomik dominant, i cili transmeton sëmundje nga brezi në brez.
Me këtë lloj trashëgimie tek një fëmijë i sëmurë, njëri nga prindërit vuan nga e njëjta sëmundje. Megjithatë, nëse vetëm njëri prind në një familje është i sëmurë dhe tjetri ka gjene të shëndetshme, atëherë fëmijët mund të mos trashëgojnë gjenin mutant.
Një shembull i trashëgimisë sipas një tipi autosomik dominant Lloji i trashëgimisë autosomale dominante mund të transmetojë më shumë se 500 patologji të ndryshme, ndër to: sindroma Marfan, sindroma Ehlers-Danlos, distrofia, sëmundja Recklinghuisen, sëmundja Huntington. Kur studiohet prejardhja, mund të gjurmohet lloji autosomal dominant i trashëgimisë.
Mund të ketë shembuj të ndryshëm për këtë, por më i habitshmi është sëmundja e Huntingtonit. Karakterizohet nga ndryshime patologjike në qelizat nervore në strukturat e trurit të përparmë.
E rëndësishme
Mozaicizmi vërehet në shumë sëmundje kromozomale. Besohet se mutacionet somatike dhe mozaicizmi luajnë një rol të rëndësishëm në etiologjinë e shumë llojeve të neoplazmave malinje.
Mozaicizmi shfaqet gjithashtu midis qelizave germinale. Gjatë oogjenezës, ndodhin 28-30 ndarje mitotike, dhe gjatë spermatogjenezës - deri në disa qindra. Në këtë drejtim, me mozaicizmin josomatik, rritet shpeshtësia e shfaqjes së mutacionit dhe rreziku i transmetimit të tij në brezat e ardhshëm.Mozaicizmi josomatik vërehet në osteogenesis imperfecta dhe disa sëmundje të trashëguara të lidhura me kromozomin X. 2. Sëmundjet mitokondriale. Mitokondritë kanë ADN-në e tyre; mtADNA ndodhet në matricën e organelës dhe përfaqësohet nga një kromozom rrethor.
Besohet se gjatë ndarjes së qelizave, mitokondritë shpërndahen rastësisht midis qelizave bija.
Kriteret për lloje të ndryshme të trashëgimisë
A. Trashëgimia monogjenike. Një tipar i koduar nga një gjen trashëgohet në përputhje me ligjet e Mendelit dhe quhet Mendelian. Tërësia e të gjitha gjeneve të një organizmi quhet gjenotip.
Një fenotip është realizimi i një gjenotipi (në aspektin morfologjik dhe biokimik) në kushte specifike mjedisore. 1. Një nga gjendjet e mundshme strukturore të një gjeni quhet alele.
Alelet lindin si rezultat i mutacioneve. Numri i mundshëm i aleleve për çdo gjen është praktikisht i pakufizuar. Në organizmat diploide, një gjen mund të përfaqësohet nga vetëm dy alele, të lokalizuara në seksione identike të kromozomeve homologe.
Gjendja kur kromozomet homologe mbajnë alele të ndryshme të të njëjtit gjen quhet heterozigot. 2. Trashëgimia e sëmundjeve monogjenike - autosomale ose të lidhura me X - mund të përcaktohet duke studiuar origjinën.
Llojet e trashëgimisë së sëmundjeve
Sëmundjet autosomale dominante karakterizohen nga një sërë shenjash të mëposhtme, të cilat zbulohen në shumicën e rasteve: 1) sëmundja transmetohet vertikalisht përgjatë origjinës, dhe rastet e sëmundjes diagnostikohen në çdo brez, 2) rreziku i trashëgimisë së sëmundjes. për cilindo nga fëmijët e pacientit është 50%; 3) anëtarët fenotipikisht normalë të familjes nuk trashëgojnë sëmundje tek pasardhësit e tyre; 4) meshkujt dhe femrat preken me frekuencë të barabartë: 5) një pjesë e konsiderueshme e rasteve të sëmundjes shkaktohen nga një mutacion i ri. Me një lloj trashëgimie autosomale dominante, ndryshe nga tipi i lidhur me X, transmetimi i sëmundjes përmes linjës mashkullore (nga babai te djali) është i mundur (Fig.
29.2). Meqenëse një burrë kalon një kromozom Y dhe jo një kromozom X tek djemtë e tij, në rastet kur një sëmundje trashëgimore kalon nga babai te djali, trashëgimia e lidhur me X është e përjashtuar.
Lajmet e seksioneve të faqes në internet
Anëtarët e shëndetshëm të familjes lindin fëmijë të shëndetshëm. 4) Sëmundjet autosomale dominante janë gjithmonë të trashëguara, pavarësisht nga gjinia e fëmijës dhe gjinia e prindit të prekur. Përjashtimet ndodhin në rastet e mutacioneve të reja dhe depërtimit jo të plotë të gjeneve.
b. Trashëgimia autosomale recesive. Sëmundjet me një model të trashëgimisë autosomale recesive përfshijnë sëmundjen Tay-Sachs, fibrozën cistike dhe shumicën e çrregullimeve metabolike të trashëguara. Sëmundjet autosomale recesive janë zakonisht më të rënda se sëmundjet autosomale dominante.
1) Nëse të dy prindërit janë të shëndetshëm, por janë bartës të një gjeni patologjik, rreziku për të pasur një fëmijë të sëmurë është 25%. 2) Një fëmijë i shëndetshëm në 2/3 e rasteve rezulton të jetë bartës heterozigot i gjenit patologjik. 3) Në një fëmijë me sëmundje autosomale recesive, veçanërisht të rrallë, prindërit shpesh rezultojnë të jenë të afërm gjaku.
e ndaluar
Meqenëse te një prind i sëmurë gjeni mutant lokalizohet në gjysmën e gameteve, të cilat mund të fekondohen në mënyrë të barabartë me qelizat normale, probabiliteti që sëmundja të shfaqet tek fëmijët është 50%. Sidoqoftë, kur analizoni origjinën, është e nevojshme të mbani mend mundësinë e depërtimit jo të plotë të alelit dominues për shkak të ndërveprimit të gjeneve ose faktorëve mjedisorë.
Të gjithë fëmijët fenotipikisht të shëndetshëm do të jenë gjenetikisht të shëndetshëm nëse depërtimi i gjenit mutant është i plotë. Në rastet e penetrencës së ulët, shenjat patologjike nuk shfaqen në disa breza.
Gjithashtu duhet theksuar se disa sëmundje nuk shfaqen që në momentin e lindjes, por vetëm në një moshë të caktuar. Kjo krijon disa vështirësi në përcaktimin e llojit të trashëgimisë.
Lloji autosomik recesiv i trashëgimisë së tiparit transmetohet
Një shembull i një sëmundjeje recesive të lidhur me X është hemofilia A, e karakterizuar nga një çrregullim i koagulimit të gjakut për shkak të mungesës së faktorit VIII - globulinës antihemofilike A. Pedigreja e një pacienti me hemofili është paraqitur në Fig. IX. 11. Klinikisht sëmundja manifestohet me gjakderdhje të shpeshta të zgjatur edhe me plagë të lehtë, hemorragji në organe dhe inde. Incidenca e sëmundjes është 1 në 10,000 djem të porsalindur.
Duke përdorur shënimet e mësipërme, është e mundur të përcaktohen të gjitha gjenotipet e mundshme në pasardhësit e një burri të sëmurë dhe një gruaje të shëndoshë (Fig. IX. 12). Sipas skemës, të gjithë fëmijët do të jenë fenotipikisht të shëndetshëm, por gjenotipisht të gjitha vajzat janë bartëse të gjenit të hemofilisë.
Nëse një grua, bartëse e gjenit të hemofilisë, martohet me një burrë të shëndetshëm, opsionet e mëposhtme për gjenotipet e pasardhësve janë të mundshme (Fig. IX. 13).
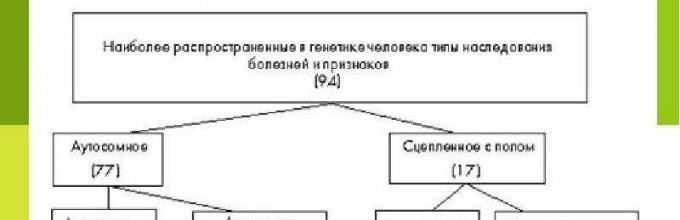
Lloji i trashëgimisë zakonisht i referohet trashëgimisë së një tipari të veçantë në varësi të faktit nëse gjeni (aleli) që e përcakton atë ndodhet në kromozomin autosomik ose seksual, dhe nëse ai është dominant ose recesiv. Në këtë drejtim, dallohen këto lloje kryesore të trashëgimisë: 1) autosomike dominante, 2) autosomale recesive, 3) trashëgimi dominante e lidhur me seksin dhe 3) trashëgimi recesive e lidhur me seksin. Prej tyre dallohen veçmas 4) llojet autosomale të kufizuara nga seksi dhe 5) llojet holandrike të trashëgimisë. Përveç kësaj, ka 6) trashëgimi mitokondriale.
Në mënyra autosomale dominante e trashëgimisë aleli i gjenit që përcakton tiparin ndodhet në një nga autozomet (kromozomet jo seksuale) dhe është dominues. Kjo simptomë do të shfaqet në të gjitha brezat. Edhe kur kryqëzohen gjenotipet Aa dhe aa, do të vërehet në gjysmën e pasardhësve.
Kur tip autosomik recesiv një tipar mund të mos shfaqet në disa breza, por të shfaqet në të tjera. Nëse prindërit janë heterozigotë (Aa), atëherë ata janë bartës të një alele recesive, por kanë një tipar dominues. Kur kryqëzohen Aa dhe Aa, ¾ e pasardhësve do të kenë një tipar mbizotërues dhe ¼ do të ketë një tipar recesiv. Kur kryqëzohen Aa dhe aa në ½, aleli recesiv i gjenit do të shfaqet në gjysmën e pasardhësve.
Tiparet autosomale ndodhin me frekuencë të barabartë në të dy gjinitë.
Trashëgimia dominuese e lidhur me seksin e ngjashme me autosomike dominuese me një ndryshim: në një seks, kromozomet seksuale të të cilit janë të njëjta (për shembull, XX në shumë kafshë është një organizëm femëror), tipari do të shfaqet dy herë më shpesh se në një seks me kromozome të ndryshme seksuale (XY). Kjo për faktin se nëse aleli i gjenit ndodhet në kromozomin X të trupit të mashkullit (dhe partneri nuk e ka fare një alele të tillë), atëherë do ta kenë të gjitha vajzat dhe asnjë nga djemtë. Nëse pronari i një tipari dominues të lidhur me seksin është një organizëm femëror, atëherë probabiliteti i transmetimit të tij është i njëjtë për të dy gjinitë e pasardhësve.
Në Mënyra recesive e trashëgimisë e lidhur me seksin Mund të ndodhë edhe kapërcimi i gjeneratave, si në rastin e tipit autosomik recesiv. Kjo vërehet kur organizmat femërorë mund të jenë heterozigotë për një gjen të caktuar, dhe organizmat mashkullorë nuk mbartin alelën recesive. Kur një bartës femër kryqëzohet me një mashkull të shëndetshëm, ½ e djemve do të shprehin gjenin recesive dhe ½ e vajzave do të jenë bartëse. Tek njerëzit, hemofilia dhe verbëria e ngjyrave trashëgohen në këtë mënyrë. Etërit nuk ua kalojnë kurrë gjenin e sëmundjes djemve të tyre (pasi ata kalojnë vetëm në kromozomin Y).
Mënyra autosomale, e kufizuar nga seksi i trashëgimisë vërehet kur gjeni që përcakton tiparin, edhe pse i lokalizuar në autosome, shfaqet vetëm në njërin nga gjinitë. Për shembull, shenja e sasisë së proteinave në qumësht shfaqet vetëm tek femrat. Nuk është aktiv tek meshkujt. Trashëgimia është afërsisht e njëjtë si në një lloj recesive të lidhur me seksin. Megjithatë, këtu tipari mund të kalohet nga babai te djali.
Trashëgimia holandezeështë e lidhur me lokalizimin e gjenit në studim në kromozomin gjinor Y. Kjo veçori, pavarësisht nëse është dominante apo recesive, do të shfaqet tek të gjithë djemtë dhe jo tek asnjë vajzë.
Mitokondritë kanë gjenomin e tyre, i cili përcakton praninë lloji i trashëgimisë mitokondriale. Meqenëse vetëm mitokondritë e vezës përfundojnë në zigot, trashëgimia mitokondriale ndodh vetëm nga nënat (si vajzat ashtu edhe djemtë).
Ligjërata: Llojet bazë të trashëgimisë së tipareve te njerëzit
Autosomike dominante lloji i trashëgimisë (Fig. 11.2) karakterizohet nga këto karakteristika:
1) pacientët në çdo gjeneratë;
2) një fëmijë i sëmurë me prindër të sëmurë;
4) trashëgimia shkon vertikalisht dhe horizontalisht;
5) probabiliteti i trashëgimisë 100%, 75% dhe 50%.
Duhet theksuar se shenjat e mësipërme të një lloji autosomik dominant të trashëgimisë do të shfaqen vetëm me dominim të plotë. Kështu trashëgohen tek njerëzit polidaktilia (këmbët me gjashtë gishta), njollat, flokët kaçurrelë, ngjyra e syve kafe, etj. Me dominim jo të plotë, hibridet do të shfaqin një formë të ndërmjetme trashëgimie. Nëse gjeni ka depërtim jo të plotë, mund të mos ketë pacientë në çdo brez.
Mënyra autosomale recesive e trashëgimisë(Fig. 11.2) karakterizohet nga karakteristikat e mëposhtme:
3) burrat dhe gratë janë të prekur në mënyrë të barabartë;
4) trashëgimia ndodh kryesisht horizontalisht;
5) probabiliteti i trashëgimisë 25%, 50% dhe 100%.
Më shpesh, probabiliteti për të trashëguar një sëmundje të një lloji autosomik recesiv është 25%, pasi për shkak të ashpërsisë së sëmundjes pacientë të tillë ose nuk mbijetojnë deri në moshën e lindjes së fëmijës.
ose mos u martua. Kështu trashëgohet tek njeriu fenilketonuria, anemia drapëroqelizore, ngjyra e syve blu etj.
Mënyra recesive e trashëgimisë e lidhur me seksin(Fig. 11.3) karakterizohet nga karakteristikat e mëposhtme:
1) pacientët nuk janë në çdo brez;
2) prindërit e shëndetshëm kanë një fëmijë të sëmurë;
3) preken kryesisht meshkujt;
4) trashëgimia ndodh kryesisht horizontalisht;
5) probabiliteti i trashëgimisë është 25% për të gjithë fëmijët dhe 50% për djemtë.
Kështu trashëgohet te njeriu hemofilia, daltonizmi, anemia trashëgimore, distrofia muskulare etj.
Mënyra dominuese e trashëgimisë e lidhur me seksin(Fig. 11.4) është e ngjashme me autosomike dominuese, me përjashtim të faktit se burri ua kalon këtë tipar të gjitha vajzave të tij (djemtë marrin një kromozom Y nga babai i tyre, ata janë të shëndetshëm). Një shembull i një sëmundjeje të tillë është një formë e veçantë e rakitave që është rezistente ndaj trajtimit me vitaminë B.
Lloji holandez i trashëgimisë(Fig. 11.5) karakterizohet nga karakteristikat e mëposhtme:
1) pacientët në të gjitha gjeneratat;
2) vetëm meshkujt sëmuren;
3) një baba i sëmurë i ka të gjithë djemtë të sëmurë;
4) probabiliteti i trashëgimisë është 100% tek djemtë.
Karakteristikat holandrike nuk janë të rëndësishme në patologjinë trashëgimore njerëzore. Sipas tipit holandrik, meshkujt trashëgojnë ichthyosis (rrëshqitje e lëkurës), hipertrikozë (rritje e tepruar e qimeve në veshë dhe kanalet e jashtme të dëgjimit), rrjetëzim midis gishtërinjve, etj.
Më shumë punë në biologji
Abstrakt mbi biologjinë
Llojet bazë të trashëgimisë së tipareve
Ekzistojnë shumë lloje të trashëgimisë së tipareve: direkte, indirekte dhe komplekse.
Trashëgimia e drejtpërdrejtë. në të cilat variantet e tipareve ruhen të pandryshuara nga brezi në brez - ky është lloji më i thjeshtë i trashëgimisë së tipareve. Trashëgimia e drejtpërdrejtë vërehet shpesh në bimët që riprodhohen në mënyrë vegjetative ose prodhojnë fara nëpërmjet vetëpllenimit, më rrallë gjatë riprodhimit të kafshëve (brenda së njëjtës race) ose pjalmimit të kryqëzuar në bimë (brenda të njëjtit varietet ose linjë).
– Trashëgimi direkte gjatë shumimit vegjetativ të bimëve
Shembulli 1. Trëndafilat e qytetit të Kinës karakterizohen nga lule të verdha të ndezura. Kur shumohen në mënyrë vegjetative, prerjet e kësaj varieteti prodhojnë gjithmonë bimë me lule të verdha të ndezura.
Shembulli 2. Disa lloje të shelgut të qarë karakterizohen nga fidaneve të verdha të ndritshme. Gjatë shumimit vegjetativ, prerjet e këtyre varieteteve prodhojnë gjithmonë pemë me një kurorë të qarë dhe lastarë të verdhë të ndezur.
– Trashëgimi direkte gjatë vetëpllenimit në bimë
Shembulli 1. Varietetet e bizeleve me fara jeshile dhe lule të bardha, kur vetëpjalmohen, prodhojnë gjithmonë bizele të gjelbra në pasardhësit e tyre, nga të cilat rriten bimë me lule të bardha.
Lexoni gjithashtu:
Shembulli 2. Varietetet e domates me fruta të zgjatura të verdha, kur vetëpjalmohen, prodhojnë gjithmonë fara nga të cilat rriten bimët me fruta të zgjatura të verdha.
– Trashëgimia e drejtpërdrejtë gjatë riprodhimit të kafshëve të racës së pastër dhe pjalmimi i kryqëzuar i bimëve të racës së pastër
Shembulli 1. Kur kryqëzohen lopët e racës së pastër dhe demat bardh e zi, të gjithë pasardhësit e tyre karakterizohen nga një ngjyrë bardh e zi.
Shembulli 2. Pjalmimi i kryqëzuar i bimëve të domates së pastër me fruta sferike të kuqe prodhon gjithmonë fara nga të cilat rriten bimët me fruta sferike të kuqe.
Trashëgimia indirekte- Kjo është një lloj trashëgimie më komplekse, e cila vërehet gjatë riprodhimit të kafshëve dhe riprodhimit të farës në bimë (që në thelb është edhe seksual). Për të studiuar trashëgiminë indirekte, është i nevojshëm hibridizimi - kalimi i organizmave që ndryshojnë në gjenotip. Me trashëgimi indirekte, disa variante tiparesh shfaqen në secilën gjeneratë (tipare të tilla quhen dominuese. "dominant"), dhe opsionet e tjera mund të "zhduken" përkohësisht dhe më pas të shfaqen në gjeneratat pasuese (karakteristika të tilla quhen recesive. "tërheqje")
Shembulli 1. Kina e lashtë është vendlindja e peshqve të artë dekorativë me një larmi ngjyrash, gjatësi fin dhe forma trupore. Peshku i kuq (si dhe krapi) janë një objekt i përshtatshëm për të demonstruar kryqëzimin: ata kanë fekondim të jashtëm, dhe gametet (pjelljet dhe qumështi) janë drejtpërdrejt të dukshme. Mijëra vjet më parë, u vu re se pasardhësit e peshqve me ngjyrë të zbehtë mund të prodhonin individë me ngjyra të artë, portokalli, të zeza dhe të larmishme. Kur kryqëzonin individë të shurdhër dhe me ngjyra të ndezura me njëri-tjetrin, në disa raste të gjithë pasardhësit e tyre kishin një ngjyrë të shurdhër - kjo është një tipar mbizotërues. Megjithatë, kur këta pasardhës u kryqëzuan me njëri-tjetrin në brezat pasardhës, individët me tipare recesive të "zhdukura" më parë u rishfaqën.
Shembulli 2. Në Japoninë mesjetare, minjtë dekorativë me ngjyra të pazakonta ishin të njohura: të bardhë, të verdhë, të zinj, me njolla. Kur minjtë e bardhë dhe të zinj u kryqëzuan me njëri-tjetrin, në disa raste të gjithë pasardhësit e tyre ishin të zinj (ngjyra e bardhë recesive u shfaq vetëm në gjeneratat pasuese), dhe në raste të tjera - e bardha (tani ngjyra e zezë ishte recesive). Vetëm në shekullin e 20-të u vërtetua se në raste të ndryshme ngjyra e bardhë përcaktohej nga gjene të ndryshme.
Shembulli 3. Kur kryqëzohen shumë bimë zbukuruese (snapdragon, bukuria e natës) me lule të kuqe dhe të bardha, bimët me një ngjyrë rozë të ndërmjetme rriten nga farat hibride. Megjithatë, kur këto bimë hibride me lule rozë kryqëzohen me njëra-tjetrën, në pasardhësit e tyre shfaqen bimë me lule të kuqe, të bardha dhe rozë.
Llojet komplekse trashëgimia e tipareve quhet komplekse sepse është shumë e vështirë të parashikohet paraprakisht shfaqja e varianteve të reja të tipareve. Në disa raste, shfaqen “papritmas” variante të reja karakteristikash që nuk i kishin as prindërit, as gjyshërit, as hallat dhe dajat. Ndonjëherë një shfaqje e tillë "e papritur" e simptomave quhet krejtësisht e paarsyeshme një mutacion.
Shembulli 1. Peshqit e bishtit të shpatës së akuariumit (dhe një grup afër bishtit të shpatës - pllaka) karakterizohen nga një larmi ngjyrash: gri-jeshile, e kuqe e errët (tulla), e kuqe e ndezur (skarlat), limon (e verdhë e lehtë), me njolla (tigër dhe basme). Këta peshq janë një lëndë e përshtatshme për demonstrim të kryqëzimit, pasi kanë fekondim të brendshëm, dhe femrat lindin të skuqura të gjalla. Kur kryqëzohen femrat e kuqe të kuqe të pastër me meshkuj të kuqe të errët të racës së pastër, gjithmonë fitohen hibride të gjelbërta-gri. Megjithatë, kur këto hibride kryqëzohen me njëri-tjetrin, pasardhësit e tyre prodhojnë individë me një larmi ngjyrash, duke përfshirë të verdhën e limonit, të cilën të gjithë paraardhësit e njohur nuk e kishin.
Shembulli 2. Shumë ushqime (fruta, kokrra të kuqe) dhe bimë zbukuruese shumohen në mënyrë vegjetative. Në të njëjtën kohë, për dekada, çdo varietet ruan karakteristikat e veta. Nëse mbledhni fara nga një bimë e tillë dhe i mbillni ato, atëherë këto fara do të rriten në bimë me kombinimet më fantastike të karakteristikave.
Lloji i trashëgimisë është autosomik dominant. Llojet e trashëgimisë së tipareve te njerëzit

Të gjitha tiparet karakteristike të trupit tonë manifestohen nën ndikimin e gjeneve. Ndonjëherë vetëm një gjen është përgjegjës për këtë, por më shpesh ndodh që disa njësi të trashëgimisë janë përgjegjëse për manifestimin e një tipari të veçantë.
Tashmë është vërtetuar shkencërisht se për një person, manifestimi i karakteristikave të tilla si ngjyra e lëkurës, flokët, sytë dhe shkalla e zhvillimit mendor varet nga aktiviteti i shumë gjeneve menjëherë. Kjo trashëgimi nuk u bindet saktësisht ligjeve të Mendelit, por shkon shumë përtej saj.
Studimi i gjenetikës njerëzore nuk është vetëm interesant, por edhe i rëndësishëm nga pikëpamja e të kuptuarit të trashëgimisë së sëmundjeve të ndryshme trashëgimore. Në ditët e sotme, po bëhet mjaft e rëndësishme që çiftet e reja të kërkojnë këshillim gjenetik, në mënyrë që pas analizimit të origjinës së secilit bashkëshort, të mund të thuhet me siguri se fëmija do të lindë i shëndetshëm.
Llojet e trashëgimisë së tipareve te njerëzit

Nëse e dini se si trashëgohet një tipar i veçantë, mund të parashikoni mundësinë e shfaqjes së tij tek pasardhësit. Të gjitha tiparet në trup mund të ndahen në dominante dhe recesive. Ndërveprimi mes tyre nuk është aq i thjeshtë dhe ndonjëherë nuk mjafton të dish se cilës kategori i përket.
Tani në botën shkencore ekzistojnë llojet e mëposhtme të trashëgimisë tek njerëzit:
- Trashëgimia monogjene.
- Poligjenike.
- Jokonvencionale.
Këto lloje të trashëgimisë, nga ana tjetër, ndahen gjithashtu në varietete të caktuara.
Trashëgimia monogjenike bazohet në ligjet e para dhe të dyta të Mendelit. Polygenic bazohet në ligjin e tretë. Kjo nënkupton trashëgiminë e disa gjeneve, më së shpeshti jo alelike.
Trashëgimia jotradicionale nuk u bindet ligjeve të trashëgimisë dhe kryhet sipas rregullave të veta, të panjohura për askënd.
Trashëgimia monogjene

Ky lloj i trashëgimisë së tipareve te njerëzit i bindet ligjeve të Mendelejevit. Duke marrë parasysh faktin se ka dy alele të secilit gjen në gjenotip, ndërveprimi midis gjenomit femëror dhe mashkullor konsiderohet veçmas për çdo çift.
Bazuar në këtë, dallohen llojet e mëposhtme të trashëgimisë:
- Autosomike dominante.
- Autosomale recesive.
- Trashëgimia dominante e lidhur me X.
- Recesiv i lidhur me X.
- Trashëgimia holandrike.
Çdo lloj trashëgimie ka karakteristikat dhe karakteristikat e veta.
Shenjat e trashëgimisë autosomale dominante

Lloji autosomik dominant i trashëgimisë është trashëgimia e tipareve mbizotëruese që ndodhen në autozome. Manifestimet e tyre fenotipike mund të ndryshojnë shumë. Për disa, simptoma mund të jetë mezi e dukshme, dhe ndonjëherë manifestimi i saj është shumë intensiv.
Lloji autosomik dominant i trashëgimisë ka karakteristikat e mëposhtme:
- Simptoma e sëmundjes shfaqet në çdo brez.
- Numri i njerëzve të sëmurë dhe të shëndetshëm është afërsisht i njëjtë, raporti i tyre është 1:1.
- Nëse fëmijët e prindërve të sëmurë lindin të shëndetshëm, atëherë fëmijët e tyre do të jenë të shëndetshëm.
- Sëmundja prek njëlloj si djemtë ashtu edhe vajzat.
- Sëmundja transmetohet në mënyrë të barabartë nga burrat dhe gratë.
- Sa më i fortë të jetë efekti në funksionet riprodhuese, aq më e madhe është mundësia e shfaqjes së mutacioneve të ndryshme.
- Nëse të dy prindërit janë të sëmurë, atëherë fëmija, i lindur homozigot për këtë tipar, është më i sëmurë në krahasim me një heterozigot.
Të gjitha këto karakteristika realizohen vetëm në kushtet e dominimit të plotë. Në këtë rast, vetëm prania e një gjeni dominues do të jetë e mjaftueshme për manifestimin e tiparit. Lloji autosomik dominant i trashëgimisë mund të vërehet tek njerëzit kur trashëgohen njollat, flokët kaçurrelë, sytë kafe dhe shumë të tjera.
Tiparet autosomale dominante

Shumica e njerëzve që janë bartës të një tipari patologjik autosomik dominant janë heterozigotë për të. Studime të shumta konfirmojnë se homozigotët për një anomali dominuese kanë manifestime më të rënda dhe më të rënda në krahasim me heterozigotët.
Lexoni gjithashtu: Kontrata për blerjen dhe shitjen e një makine me trashëgimi - mostër
Kjo lloj trashëgimie tek njerëzit është karakteristikë jo vetëm për tipare patologjike, por edhe disa krejtësisht normale trashëgohen në këtë mënyrë.
Ndër karakteristikat normale me këtë lloj trashëgimie janë:
- Floke kacurrela.
- Sytë e errët.
- Hundë e drejtë.
- Një gunga në urën e hundës.
- Tullaciteti në moshë të hershme tek meshkujt.
- Djathtasja.
- Aftësia për të rrotulluar gjuhën në një tub.
- Gropë në mjekër.
Ndër anomalitë që kanë një mënyrë autosomale dominuese të trashëgimisë, më të famshmet janë këto:
- Me shumë gishta, mund të jetë në të dy duart dhe këmbët.
- Shkrirja e indeve të falangave të gishtave.
- Brakidaktilia.
- sindromi Marfan.
- Miopia.
Nëse dominimi është i paplotë, atëherë manifestimi i tiparit nuk mund të vërehet në çdo brez.
Mënyra autosomale recesive e trashëgimisë

Një tipar me këtë lloj trashëgimie mund të shfaqet vetëm nëse formohet një homozigot për këtë patologji. Sëmundje të tilla janë më të rënda, sepse të dy alelet e një gjeni janë me defekt.
Mundësia e shfaqjes së shenjave të tilla rritet në martesat e lidhura ngushtë, prandaj në shumë vende është e ndaluar të hyni në një aleancë midis të afërmve.
Kriteret kryesore për një trashëgimi të tillë përfshijnë si më poshtë:
- Nëse të dy prindërit janë të shëndetshëm, por janë bartës të një gjeni patologjik, atëherë fëmija do të jetë i sëmurë.
- Gjinia e fëmijës së palindur nuk luan ndonjë rol në trashëgimi.
- Për një çift të martuar, rreziku për të pasur një fëmijë të dytë me të njëjtën patologji është 25%.
- Nëse shikoni origjinën, mund të shihni një shpërndarje horizontale të pacientëve.
- Nëse të dy prindërit janë të sëmurë, atëherë të gjithë fëmijët do të lindin me të njëjtën patologji.
- Nëse njëri prind është i sëmurë dhe tjetri është bartës i një gjeni të tillë, atëherë probabiliteti për të pasur një fëmijë të sëmurë është 50%.
Shumë sëmundje metabolike trashëgohen sipas këtij lloji.
Lloji i trashëgimisë i lidhur me kromozomin X

Kjo trashëgimi mund të jetë ose dominante ose recesive. Shenjat e trashëgimisë dominuese përfshijnë si më poshtë:
- Mund të preken të dyja gjinitë, por femrat janë 2 herë më të predispozuara.
- Nëse një baba është i sëmurë, atëherë ai mund t'ua kalojë gjenin e sëmurë vetëm vajzave të tij, sepse djemtë marrin kromozomin Y prej tij.
- Një nënë e sëmurë ka të njëjtat gjasa t'ua japë këtë sëmundje fëmijëve të të dy gjinive.
- Sëmundja është më e rëndë te meshkujt sepse u mungon kromozomi i dytë X.
Nëse ka një gjen recesiv në kromozomin X, atëherë trashëgimia ka karakteristikat e mëposhtme:
- Një fëmijë i sëmurë mund të lindë edhe nga prindër fenotipikisht të shëndetshëm.
- Më së shpeshti preken meshkujt dhe femrat janë bartëse të gjenit të sëmurë.
- Nëse babai është i sëmurë, atëherë nuk duhet të shqetësoheni për shëndetin e djemve tuaj, ata nuk mund të marrin një gjen të dëmtuar prej tij.
- Probabiliteti për të lindur një fëmijë të sëmurë në një grua bartëse është 25% nëse flasim për djem, atëherë rritet në 50%.
Kështu trashëgohen sëmundje si hemofilia, verbëria e ngjyrave, distrofia muskulare, sindroma Kallmann e disa të tjera.
Sëmundjet autosomale dominante

Për shfaqjen e sëmundjeve të tilla mjafton prania e një gjeni me defekt, nëse ai është dominant. Sëmundjet autosomale dominante kanë disa karakteristika:
- Aktualisht numërohen rreth 4000 mijë sëmundje të tilla.
- Të dyja gjinitë preken në mënyrë të barabartë.
- Shfaqet qartë demorfizmi fenotipik.
- Nëse një mutacion i një gjeni mbizotërues ndodh në gamete, ai ka shumë të ngjarë të shfaqet në gjeneratën e parë. Tashmë është vërtetuar se meshkujt kanë një rrezik në rritje për të marrë mutacione të tilla me kalimin e moshës, që do të thotë se ata mund t'u japin fëmijëve të tyre sëmundje të tilla.
- Sëmundja shpesh manifestohet në të gjitha brezat.
Trashëgimia e një gjeni me defekt për një sëmundje autosomale dominante nuk ka të bëjë fare me gjininë e fëmijës apo shkallën e zhvillimit të kësaj sëmundjeje tek prindi.
Sëmundjet autosomale dominante përfshijnë:
- sindromi Marfan.
- Sëmundja e Huntingtonit.
- Neurofibromatoza.
- Skleroza tuberoze.
- Sëmundja e veshkave policistike dhe shumë të tjera.
Të gjitha këto sëmundje mund të manifestohen në shkallë të ndryshme në pacientë të ndryshëm.
sindromi Marfan

Kjo sëmundje karakterizohet nga dëmtimi i indit lidhës, dhe për rrjedhojë funksionimi i tij. Gjymtyrët e gjata në mënyrë disproporcionale me gishta të hollë sugjerojnë sindromën Marfan. Lloji i trashëgimisë së kësaj sëmundjeje është autosomik dominant.
Simptomat e mëposhtme të kësaj sindrome mund të renditen:
- Ndërtim i hollë.
- Gishtat e gjatë "merimangë".
- Defektet e sistemit kardiovaskular.
- Shfaqja e strijave në lëkurë pa ndonjë arsye të dukshme.
- Disa pacientë raportojnë dhimbje në muskuj dhe kocka.
- Zhvillimi i hershëm i osteoartritit.
- Rachiocampsis.
- Lidhje shumë fleksibël.
- Dëmtime të mundshme të të folurit.
- Dëmtimi i shikimit.
Simptomat e kësaj sëmundjeje mund t'i emërtoni për një kohë të gjatë, por shumica e tyre lidhen me sistemin skeletor. Diagnoza përfundimtare do të bëhet pasi të jenë përfunduar të gjitha ekzaminimet dhe të gjenden shenjat karakteristike në të paktën tre sisteme organesh.
Mund të vërehet se për disa, shenjat e sëmundjes nuk shfaqen në fëmijëri, por bëhen të dukshme disi më vonë.
Edhe tani, kur niveli i mjekësisë është mjaft i lartë, është e pamundur të kurohet plotësisht sindroma Marfan. Duke përdorur barnat dhe teknologjitë moderne të trajtimit, është e mundur të zgjatet jeta e pacientëve me këtë çrregullim dhe të përmirësohet cilësia e tij.
Aspekti më i rëndësishëm i trajtimit është parandalimi i zhvillimit të aneurizmës së aortës. Kërkohen konsulta të rregullta me një kardiolog. Në raste urgjente, indikohet kirurgjia e transplantit të aortës.
Korea e Huntingtonit

Kjo sëmundje ka gjithashtu një mënyrë trashëgimie autosomale dominante. Fillon të shfaqet nga mosha 35-50 vjeç. Kjo është për shkak të vdekjes progresive të neuroneve. Klinikisht, shenjat e mëposhtme mund të identifikohen:
- Lëvizjet e çrregullta të kombinuara me ulje të tonit.
- Sjellja antisociale.
- Apati dhe nervozizëm.
- Manifestimi i tipit skizofrenik.
- Luhatje humori.
Trajtimi synon vetëm eliminimin ose zvogëlimin e simptomave. Ata përdorin qetësues dhe neuroleptikë. Asnjë trajtim nuk mund të ndalojë zhvillimin e sëmundjes, kështu që vdekja ndodh afërsisht 15-17 vjet pas shfaqjes së simptomave të para.
Trashëgimia poligjenike

Shumë shenja dhe sëmundje kanë një mënyrë trashëgimie autosomale dominante. Se çfarë është kjo tashmë është e qartë, por në shumicën e rasteve nuk është aq e thjeshtë. Shumë shpesh, jo një, por disa gjene trashëgohen njëkohësisht. Ato manifestohen në kushte specifike mjedisore.
Një tipar dallues i kësaj trashëgimie është aftësia për të rritur efektin individual të secilit gjen. Karakteristikat kryesore të një trashëgimie të tillë përfshijnë si më poshtë:
- Sa më e rëndë të jetë sëmundja, aq më i madh është rreziku i zhvillimit të kësaj sëmundje tek të afërmit.
- Shumë tipare multifaktoriale ndikojnë në një gjini të caktuar.
- Sa më shumë të afërm që e kanë këtë tipar, aq më i lartë është rreziku i zhvillimit të kësaj sëmundje tek pasardhësit e ardhshëm.
Të gjitha llojet e konsideruara të trashëgimisë i përkasin varianteve klasike, por, për fat të keq, shumë shenja dhe sëmundje nuk mund të shpjegohen sepse i përkasin trashëgimisë jo tradicionale.
Kur planifikoni lindjen e një fëmije, mos e neglizhoni vizitën e një konsulte gjenetike. Një specialist kompetent do t'ju ndihmojë të kuptoni origjinën tuaj dhe të vlerësoni rrezikun e lindjes së një fëmije me aftësi të kufizuara.
Gabimet e pafalshme të filmave që ndoshta nuk i keni vënë re kurrë Ka ndoshta shumë pak njerëz që nuk u pëlqen të shikojnë filma. Megjithatë, edhe në kinemanë më të mirë ka gabime që shikuesi mund t'i vërë re.
15 gratë më të bukura të milionerëve Shikoni listën e grave të njerëzve më të suksesshëm në botë. Ata janë bukuroshe mahnitëse dhe shpesh janë të suksesshme në biznes.
Pse nuk keni parë kurrë një pëllumb të vogël? Shkoni në çdo shesh të qytetit dhe, pa dyshim, do të shihni qindra pëllumba që fluturojnë rreth kalimtarëve. Por, përkundër një numri kaq të madh.
LLOJI TRASHËGIMISË AUTOSOMAL DOMINANT
Shembuj të sëmundjeve: Sindroma Marfan, hemoglobinopatia M, korea e Huntingtonit, polipoza e zorrës së trashë, hiperkolesterolemia familjare, neurofibromatoza, polidaktilia.
Lloji autosomik dominant i trashëgimisë karakterizohet nga sa vijon shenjat:
· Frekuencë e barabartë e patologjisë tek meshkujt dhe femrat.
· Prania e pacientëve në çdo gjeneratë të pedigresë, d.m.th. transmetimi i rregullt i sëmundjes nga brezi në brez (e ashtuquajtura shpërndarja vertikale e sëmundjes).
· Probabiliteti për të pasur një fëmijë të sëmurë është 50% (pavarësisht gjinisë së fëmijës dhe numrit të lindjeve).
· Anëtarët e paprekur të familjes, si rregull, kanë pasardhës të shëndetshëm (pasi nuk kanë gjenin mutant).
Karakteristikat e listuara realizohen sipas kushtit dominim i plotë(prania e një gjeni dominues është e mjaftueshme për zhvillimin e një pasqyre specifike klinike të sëmundjes). Kështu trashëgohen tek njerëzit njollat, flokët kaçurrelë, ngjyra e syve kafe, etj. Nëse gjeni ka depërtim jo të plotë, mund të mos ketë pacientë në çdo brez.
LLOJI I TRASHËGIMISË AUTOSOMAL RECESIV
Shembuj të sëmundjeve: fenilketonuria, albinizmi okular-lëkuror, anemia drapërocitare, sindroma adrenogenitale, galaktosemia, glikogjenoza, hiperlipoproteinemia, fibroza cistike.
Mënyra autosomale recesive e trashëgimisë karakterizohet nga sa vijon shenjat:
· Frekuencë e barabartë e patologjisë tek meshkujt dhe femrat.
· Manifestimi i patologjisë në origjinë “horizontalisht”, shpesh tek vëllezërit e motrat.
· Mungesa e sëmundjes në gjysëm gjak (fëmijë të të njëjtit baba nga nëna të ndryshme) dhe gjysmë vëllezër (fëmijë të së njëjtës nënë nga baballarë të ndryshëm).
· Prindërit e pacientit janë zakonisht të shëndetshëm. E njëjta sëmundje mund të zbulohet te të afërmit e tjerë, për shembull, te kushërinjtë ose kushërinjtë e dytë të pacientit.
Shfaqja e patologjisë autosomale recesive është më e mundshme në martesat familjare për shkak të gjasave më të mëdha për të takuar dy bashkëshortë që janë heterozigotë për të njëjtën alele patologjike të marrë nga paraardhësi i tyre i përbashkët. Sa më e madhe të jetë shkalla e marrëdhënies mes bashkëshortëve, aq më e lartë është kjo probabilitet. Më shpesh, probabiliteti për të trashëguar një sëmundje të një lloji autosomik recesiv është 25%, pasi për shkak të ashpërsisë së sëmundjes, pacientë të tillë ose nuk jetojnë deri në moshën e lindjes ose nuk martohen.
TRASHËGIMIA X-DOMINANTE E LIDHUR ME KROMOZOMET
Shembuj të sëmundjeve: një formë e hipofosfatemisë është rakitizmi rezistent ndaj vitaminës D; Sëmundja Charcot-Marie-Tooth - dominante e lidhur; Sindroma orofacialo-dixhitale e tipit I.
Shenjat e sëmundjes:
· Meshkujt dhe femrat janë të prekur, por femrat janë 2 herë më të rrezikuara.
· Transferimi i një alele patologjike nga një i sëmurë tek të gjitha vajzat dhe vetëm tek vajzat, por jo tek djemtë. Djemtë marrin kromozomin Y nga babai i tyre.
· Transmetimi i sëmundjes nga një grua e sëmurë tek djemtë dhe vajzat është po aq i mundshëm.
· Sëmundja është më e rëndë te meshkujt sesa te femrat.
TRASHËGIMIA RECESIVE E LIDHUR ME X
Shembuj të sëmundjeve: hemofilia A, hemofilia B; Sëmundja recesive Charcot-Marie-Tooth e lidhur me X; verbëria e ngjyrave; Distrofia muskulare Duchenne-Becker; sindromi Kallmann; Sëmundja e Hunterit (mukopolisakaridoza e tipit II); hipogamaglobulinemia e tipit Brutonian.
Shenjat e sëmundjes:
· Pacientët lindin nga prindër fenotipikisht të shëndetshëm.
· Sëmundja shfaqet pothuajse ekskluzivisht tek meshkujt. Nënat e pacientëve janë bartëse të detyrueshme të gjenit patologjik.
· Një djalë nuk trashëgon kurrë një sëmundje nga babai i tij.
· Një bartës i një gjeni mutant ka një shans 25% për të pasur një fëmijë të sëmurë (pavarësisht gjinisë së të porsalindurit); probabiliteti për të pasur një djalë të sëmurë është 50%.
HOLANDRIKE, OSE E LIDHUR ME KROMOZOMIN Y,
LLOJI I TRASHËGIMISË
Shembuj të shenjave: ihtioza e lëkurës, hipertrikoza e veshkave, rritja e tepërt e qimeve në falangat e mesme të gishtave, azoospermia.
Shenjat:
· Transferimi i një tipari nga babai tek të gjithë djemtë dhe djemtë e vetëm.
· Vajzat nuk trashëgojnë kurrë një tipar nga babai i tyre.
· Natyra “vertikale” e trashëgimisë së një tipari.
· Probabiliteti i trashëgimisë për meshkujt është 100%.
TRASHËGIMIA MITOKONDRIALE
Shembuj të sëmundjeve(sëmundjet mitokondriale): atrofia optike Leber, sindroma Leigh (mioencefalopatia mitokondriale), MERRF (epilepsia mioklonike), kardiomiopatia e zgjeruar familjare.
Shenjat e sëmundjes:
· Prania e patologjisë në të gjithë fëmijët e një nëne të sëmurë.
· Lindja e fëmijëve të shëndetshëm nga një baba i sëmurë dhe një nënë e shëndoshë.
Këto veçori shpjegohen me faktin se mitokondritë trashëgohen nga nëna. Pjesa e gjenomit mitokondrial atëror në zigot është ADN nga 0 deri në 4 mitokondri, dhe gjenomi i nënës është ADN nga afërsisht 2500 mitokondri. Përveç kësaj, duket se pas fekondimit, replikimi i ADN-së atërore është i bllokuar.
Me gjithë diversitetin e sëmundjeve gjenetike në patogjenezën e tyre ekziston model i përgjithshëm: lidhet fillimi i patogjenezës së ndonjë sëmundje gjenetike efekti primar i alelit mutant- një produkt primar patologjik (cilësisht ose sasior), i cili përfshihet në zinxhirin e proceseve biokimike dhe çon në formimin e defekteve në qelizore, organike Dhe nivelet e organizmit.
Patogjeneza e sëmundjes në nivel molekular shpaloset në varësi të natyrës së produktit të gjenit mutant në formën e çrregullimeve të mëposhtme:
Sinteza jonormale e proteinave;
Mungesa e prodhimit të produktit primar (më e zakonshme);
Prodhimi i një sasie të reduktuar të produktit primar normal (në këtë rast, patogjeneza është shumë e ndryshueshme);
Prodhimi i një sasie të tepërt produkti (ky opsion supozohet vetëm, por ende nuk është zbuluar në forma specifike të sëmundjeve trashëgimore).
Opsionet për zbatimin e veprimit të një gjeni jonormal:
1) gjen jonormal → ndërprerje e sintezës së mARN → ndërprerje e sintezës së proteinave → sëmundje trashëgimore;
2) gjen jonormal → ndërprerje e sintezës së mARN → sëmundje trashëgimore;
3) një gjen jonormal me një kod patologjik → sinteza e mRNA patologjike → sinteza e proteinës patologjike → sëmundje trashëgimore;
4) ndërprerje e ndezjes dhe fikjes së gjeneve (shtypja dhe depresioni i gjeneve);
5) gjen jonormal → mungesë e sintezës së receptorit hormonal → patologji hormonale trashëgimore.
Shembuj të variantit të parë të patologjisë së gjeneve: hipoalbuminemia, afibrinogenemia, hemofilia A (faktori VIII), hemofilia B (IX - faktori i Krishtlindjeve), hemofilia C (faktori XI - Rosenthal), agamaglobulinemia.
Shembuj të opsionit të dytë: albinizmi (mungesa e enzimës - tirozinaza → depigmentimi); fenilketonuria (mungesa e fenilalaninës hidroksilazë → grumbullohet fenilalanina → produkti i saj metabolik, fenilpiruvati, është toksik për sistemin nervor qendror → zhvillohet prapambetja mendore); alkaptonuria (mungesa e oksidazës së acidit homogjentisik → acidi homogjentisik grumbullohet në gjak, urinë, inde → ngjyrosja e indeve, kërc); methemoglobinemia enzimopatike (mungesa e methemoglobin reduktazës → grumbullohet methemoglobina → zhvillohet hipoksia); sindromi adrenogenital (një nga sëmundjet trashëgimore më të zakonshme të njeriut: frekuenca në Evropë 1:5000, tek eskimezët e Alaskës 1:400 - 1:150; defekti i 21-hidroksilazës → mungesa e kortizolit, akumulimi i androgjeneve → tek burrat - zhvillimi i përshpejtuar seksual, tek femrat - virilizimi).
Shembull i variantit të tretë të patologjisë së gjeneve: M - hemoglobinoza (sintetizohet hemoglobina M jonormale, e cila ndryshon nga hemoglobina A normale në atë që në pozicionin 58 të zinxhirit α (ose në pozicionin 63 të zinxhirit β) histidina zëvendësohet nga tirozina → M-hemoglobina hyn në një lidhje e fortë me oksigjenin, duke mos e dhënë atë në inde, formon methemoglobinë → zhvillohet hipoksia).
Shembull i opsionit 4: Talasemia. Dihet se qelizat e kuqe të gjakut të fetusit përmbajnë hemoglobinë të veçantë fetale, sinteza e së cilës kontrollohet nga dy gjene. Pas lindjes, veprimi i njërit prej këtyre gjeneve frenohet dhe një gjen tjetër ndizet, duke siguruar sintezën e Hb A (95-98% e hemoglobinës te njerëzit e shëndetshëm). Në patologji, mund të vërehet vazhdimësia e sintezës së hemoglobinës fetale (sasia e saj te njerëzit e shëndetshëm është 1-2%). Hb S është më pak i qëndrueshëm se Hb A - prandaj zhvillohet anemia hemolitike.
Shembull i opsionit 5: feminizimi i testikujve. Është zbuluar se individët me këtë sëmundje nuk kanë receptorë testosteroni. Prandaj, embrioni mashkull fiton tipare karakteristike për trupin e femrës.
Patogjeneza e çdo sëmundjeje trashëgimore në individë të ndryshëm, megjithëse të ngjashme në mekanizmat dhe fazat parësore, formohet rreptësisht individualisht- procesi patologjik, i nisur nga efekti primar i alelit mutant, fiton integritet me variacionet natyrore individuale në varësi të gjenotipit të organizmit dhe kushteve të mjedisit.
Karakteristikat foto klinike sëmundjet e gjeneve shkaktohen nga parimet shprehja, shtypja dhe ndërveprimi i gjeneve.
Dallohen në vijim Karakteristikat kryesore të sëmundjeve të gjeneve:tiparet e pamjes klinike; polimorfizmi klinik; heterogjeniteti gjenetik. Në të njëjtën kohë, është e pamundur të vëzhgohen plotësisht të gjitha tiparet e përbashkëta në një sëmundje. Njohja e veçorive të përgjithshme të sëmundjeve të gjeneve do t'i lejojë mjekut të dyshojë për një sëmundje trashëgimore edhe në një rast sporadik.
Karakteristikat e pamjes klinike:
shumëllojshmëri manifestimesh- procesi patologjik prek disa organe tashmë në fazat fillestare të formimit të sëmundjes;
mosha të ndryshme të fillimit të sëmundjes;
përparimi i figurës klinike dhe ecuria kronike;
I kushtëzuar paaftësia që nga fëmijëria dhe jetëgjatësia e shkurtuar.
Shumëllojshmëria e manifestimeve dhe përfshirja e shumë organeve dhe indeve në procesin patologjik për këtë grup sëmundjesh është për faktin se defekti primar lokalizohet në strukturat qelizore dhe ndërqelizore të shumë organeve. Për shembull, në sëmundjet trashëgimore të indit lidhor, sinteza e një proteine të një ose një strukture fibroze specifike për secilën sëmundje është ndërprerë. Duke qenë se indi lidhor është i pranishëm në të gjitha organet dhe indet, shumëllojshmëria e simptomave klinike në këto sëmundje është pasojë e anomalive të indit lidhor në organe të ndryshme.
Mosha e fillimit për këtë grup sëmundjesh praktikisht i pakufizuar: nga fazat e hershme të zhvillimit embrional (defekte kongjenitale) - deri në pleqëri ( sëmundja e Alzheimerit). Baza biologjike e moshave të ndryshme të shfaqjes së sëmundjeve të gjeneve qëndron në modelet rreptësisht të përkohshme të rregullimit ontogjenetik të shprehjes së gjeneve. Arsyet për mosha të ndryshme të fillimit të së njëjtës sëmundje mund të jenë karakteristikat individuale të gjenomit të pacientit. Efekti i gjeneve të tjera në manifestimin e efektit të gjenit mutant mund të ndryshojë kohën e zhvillimit të sëmundjes. Koha e fillimit të veprimit të gjeneve patologjike dhe kushtet mjedisore janë gjithashtu të rëndësishme, veçanërisht në periudhën prenatale. Të dhënat e përgjithësuara për kohën e shfaqjes klinike të sëmundjeve të gjeneve tregojnë se 25% e të gjitha sëmundjeve të gjeneve zhvillohen në mitër dhe pothuajse 50% e sëmundjeve të gjeneve manifestohen në tre vitet e para të jetës.
Shumica e sëmundjeve të gjeneve karakterizohen nga përparimi i figurës klinike Dhe ecuri kronike e zgjatur me rikthime. Ashpërsia e sëmundjes "rritet" me zhvillimin e procesit patologjik. Baza biologjike primare Kjo karakteristikë është vazhdimësia e funksionimit të gjenit patologjik (ose mungesa e produktit të tij). Kjo shoqërohet me përmirësimin e procesit patologjik proceset dytësore: inflamacion; distrofia; çrregullime metabolike; hiperplazia.
Shumica e sëmundjeve të gjeneve janë të rënda dhe çojnë në paaftësia në fëmijëri Dhe shkurton jetëgjatësinë. Sa më i rëndësishëm të jetë një proces i përcaktuar monogjenik në sigurimin e aktivitetit jetësor, aq më i rëndë është manifestimi klinik i mutacionit.
Koncepti "polimorfizmi klinik" bashkohen:
Ndryshueshmëria: koha e fillimit të sëmundjes; ashpërsia e simptomave; kohëzgjatja e së njëjtës sëmundje;
Toleranca ndaj terapisë.
Shkaqet gjenetike të polimorfizmit klinik mund të përcaktohen jo vetëm nga gjeni patologjik, por edhe nga gjenotipi në tërësi, domethënë mjedisi gjenotipik në formën e gjeneve modifikuese. Gjenomi në tërësi funksionon si një sistem shumë i koordinuar. Së bashku me gjenin patologjik, individi trashëgon nga prindërit e tij kombinime të gjeneve të tjera që mund të rrisin ose dobësojnë efektin e gjenit patologjik. Përveç kësaj, në zhvillimin e një sëmundjeje gjenetike, si çdo tipar trashëgues, nuk ka rëndësi vetëm gjenotipi, por edhe mjedisi i jashtëm. Ka shumë dëshmi për këtë pozicion nga praktika klinike. Për shembull, simptomat e fenilketonurisë tek një fëmijë janë më të rënda nëse gjatë zhvillimit të saj para lindjes, dieta e nënës përfshinte shumë ushqime të pasura me fenilalaninë.
Ekziston një koncept heterogjeniteti gjenetik duke u maskuar si polimorfizëm klinik.
Heterogjeniteti gjenetik do të thotë se forma klinike e një sëmundjeje gjenetike mund të shkaktohet nga:
mutacione në gjene të ndryshme, enzimat koduese të një rruge metabolike;
mutacione të ndryshme në të njëjtin gjen, duke çuar në shfaqjen e aleleve të ndryshme (alele të shumëfishta).
Në fakt, në këto raste bëhet fjalë për të ndryshme forma nozologjike, nga pikëpamja etiologjike, e kombinuar në një formë për shkak të ngjashmërisë klinike të fenotipit. Fenomeni i heterogjenitetit gjenetik është i përgjithshëm, ai mund të quhet rregull, pasi vlen për të gjitha proteinat e trupit, duke përfshirë jo vetëm variantet patologjike, por edhe ato normale.
Deshifrimi i heterogjenitetit të sëmundjeve të gjeneve vazhdon intensivisht në dy drejtime:
klinike- sa më saktë të studiohet fenotip(analiza e pasqyrës klinike të sëmundjes), aq më shumë mundësi ka në zbulimin e formave të reja të sëmundjeve, në ndarjen e formës së studiuar në disa njësi nozologjike;
gjenetike- jep informacionin më të plotë për heterogjenitetin e formës klinike të sëmundjes Metoda e sondës së ADN-së(një metodë moderne për analizimin e gjeneve njerëzore). Caktimi i një gjeni në një ose grupe të ndryshme lidhëse, lokalizimi i gjenit, struktura e tij, thelbi i mutacionit - e gjithë kjo bën të mundur identifikimin e formave nozologjike.
Koncepti heterogjeniteti gjenetik i sëmundjeve të gjeneve hap shumë mundësi për të kuptuar thelbin e formave dhe shkaqeve individuale të polimorfizmit klinik, i cili është jashtëzakonisht i rëndësishëm për mjekësinë praktike dhe ofron këto mundësi: diagnozë të saktë; zgjedhja e metodës së trajtimit; këshillim mjekësor dhe gjenetik.
Kuptimi epidemiologjia e sëmundjeve të gjeneve e nevojshme për një mjek të çdo specialiteti, pasi në praktikën e tij mund të ndeshet me manifestime të një sëmundjeje të rrallë trashëgimore brenda zonës apo popullatës që shërben. Njohja e modeleve dhe mekanizmit të përhapjes së sëmundjeve të gjeneve do ta ndihmojë mjekun të zhvillojë në kohë masat parandaluese: ekzaminimi për heterogjenitet; këshillimi gjenetik.
Epidemiologjia e sëmundjeve të gjeneve përfshin informacionin e mëposhtëm:
Për prevalencën e këtyre sëmundjeve;
Rreth frekuencave të transportit heterozigot dhe faktorëve që i përcaktojnë ato.
Prevalenca e sëmundjes(ose numri i pacientëve) në popullatë përcaktuar nga modelet e popullsisë: intensiteti i procesit të mutacionit; presioni i përzgjedhjes, i cili përcakton pjellorinë e mutantëve dhe heterozigotëve në kushte specifike mjedisore; migrimi i popullsisë; izolim; zhvendosje gjenetike. Të dhënat për prevalencën e sëmundjeve trashëgimore janë ende fragmentare për arsyet e mëposhtme: një numër i madh i formave nozologjike të sëmundjeve gjenetike; rrallësia e tyre; diagnoza jo e plotë klinike dhe patologjike e patologjisë trashëgimore. Vlerësimi më objektiv i prevalencës së këtyre sëmundjeve në popullata të ndryshme është përcaktimi i numrit të tyre tek të porsalindurit, përfshirë të lindurit e vdekur. Frekuenca e përgjithshme e të porsalindurve me sëmundje gjenetike në popullatën në tërësi është afërsisht 1%, nga të cilat:
Me një lloj trashëgimie autosomale dominante - 0.5%;
Me autosomale recesive - 0,25%;
X-lidhur - 0,25%;
Sëmundjet e lidhura me Y dhe ato mitokondriale janë jashtëzakonisht të rralla.
Prevalenca e formave individuale të sëmundjes varion nga 1:500 (hemokromatoza primare) deri në 1:100000 dhe më poshtë (degjenerimi hepatolentikular, fenilketonuria).
Prevalenca e një sëmundjeje gjenetike konsiderohet:
E lartë - nëse 1 pacient shfaqet për 10,000 të porsalindur ose më shpesh;
Simbolet bazë:
Kur ndërtoni origjinë, është e nevojshme të vëzhgoni duke ndjekur rregullat :
a) është e nevojshme të zbulohet numri i brezave nga historia e mbledhur;
b) pedigreja fillon të ndërtohet nga probandi;
c) çdo brez numërohet me numra romakë në të majtë;
d) simbolet që tregojnë individë të një brezi janë të vendosura në një vijë horizontale dhe të numëruara me numra arabë.
Janë të mëposhtmet llojet e trashëgimisë shenjat:
autosomale dominante;
autosomale recesive;
X-lidhur (i lidhur me seksin) dominues;
X-lidhur (i lidhur me seksin) recesive;
holandrike.
Lloji autosomik dominant i trashëgimisë
Një fëmijë i sëmurë lind nga prindër të sëmurë me një probabilitet 100% nëse ata janë homozigotë; 75% nëse janë heterozigotë.
Mënyra autosomale recesive e trashëgimisë
Të dy burrat dhe gratë janë të prekur në mënyrë të barabartë.
Probabiliteti për të pasur një fëmijë të sëmurë nga prindër të shëndetshëm është 25% nëse janë heterozigotë, 0% nëse të dy ose njëri prej tyre është homozigot për gjenin dominues.
Shpesh manifestohet në martesat familjare.
Lloji mbizotërues i trashëgimisë i lidhur me X (i lidhur me seksin).
Të sëmurët ndodhin në çdo brez.
Femrat janë më të prekura.
Nëse një baba është i sëmurë, atëherë të gjitha vajzat e tij janë të sëmura.
Një fëmijë i sëmurë lind nga prindër të sëmurë me një probabilitet prej 100% nëse nëna është homozigote, 75% nëse nëna është heterozigote.
Probabiliteti për të pasur një fëmijë të sëmurë nga prindër të shëndetshëm është 0%.
Mënyra recesive e trashëgimisë e lidhur me X (e lidhur me seksin).
Pacientët nuk ndodhin në çdo brez.
Më së shumti janë të prekur meshkujt.
Probabiliteti për të lindur një djalë të sëmurë nga prindër të shëndetshëm është 25%, dhe një vajzë e sëmurë është 0%.
Lloji holandez i trashëgimisë
Të sëmurët ndodhin në çdo brez.
Vetëm meshkujt sëmuren.
Nëse një baba është i sëmurë, atëherë të gjithë djemtë e tij janë të sëmurë.
Probabiliteti për të lindur një djalë të sëmurë nga një baba i sëmurë është 100%.
Binjake metoda (e propozuar në 1876 nga F. Galton) është studimi i modeleve gjenetike te binjakët.
Thelbi i metodës: krahasimi i karakteristikave në grupe të ndryshme binjakësh bazuar në ngjashmëritë (përputhshmëria) ose dallimet (diskordancë) e tyre.
Hapat e metodës:
1. Përpilimi i një kampioni binjakësh nga e gjithë popullata.
2. Diagnoza e zigozitetit të binjakëve.
3. Përcaktimi i rolit relativ të trashëgimisë dhe mjedisit në formimin e një tipari.
D  Për të vlerësuar rolin e trashëgimisë dhe mjedisit në formimin dhe zhvillimin e një tipari, ata përdorin Formula e Holzinger-it:
Për të vlerësuar rolin e trashëgimisë dhe mjedisit në formimin dhe zhvillimin e një tipari, ata përdorin Formula e Holzinger-it:
ku N është proporcioni i faktorëve trashëgues, përputhshmëria KMB% dhe KDB% e binjakëve monozigotikë dhe dizigotikë si përqindje.
Nëse H është më i madh se 0,5, atëherë gjenotipi luan një rol të madh në formimin e tiparit nëse H është më i vogël se 0,5, atëherë mjedisi luan një rol të madh.
Metoda citogjenetike - Ky është studimi i një kariotipi duke përdorur teknologjinë mikroskopike.
Hapat e metodës:
1. Marrja dhe kultivimi i qelizave (limfocitet, fibroblaste) në mjedise ushqyese artificiale.
2. Shtimi i fitohemaglutininës në mediumin ushqyes për të stimuluar ndarjen e qelizave.
3. Ndalimi i ndarjes qelizore në fazën e metafazës duke shtuar kolkicinën.
4. Trajtimi i qelizave me tretësirë hipotonike NaCl, si rezultat i së cilës shkatërrohet membrana qelizore dhe fitohet një “shpërndarje” e kromozomeve.
5. Ngjyrosja e kromozomeve me ngjyra specifike.
6. Mikroskopimi dhe fotografimi i kromozomeve.
7. Hartimi i idiogramit dhe analiza e tij.
Metoda lejon:
a) të diagnostikojë mutacionet gjenomike dhe kromozomale;
b) të përcaktojë seksin gjenetik të organizmit.
Metoda statistikore e popullsisë -është studimi i përbërjes gjenetike të një popullate. Ai bazohet në ligjin Hardy-Weinberg dhe na lejon të përcaktojmë frekuencën e gjeneve dhe gjenotipeve në popullata të ndryshme (shih seksionin VII).
Metodat biokimike. Shkaku i shumicës së sëmundjeve monogjene trashëgimore janë defektet metabolike të shoqëruara me enzimopati (çrregullime në strukturën e enzimave të përfshira në reaksionet metabolike). Në të njëjtën kohë, produktet metabolike të ndërmjetme grumbullohen në trup, dhe për këtë arsye, duke i përcaktuar ato ose aktivitetin e enzimave, duke përdorur metoda biokimike, është e mundur të diagnostikohen sëmundjet trashëgimore metabolike (mutacionet e gjeneve). Metodat sasiore biokimike (testet e stresit) bëjnë të mundur identifikimin e transportit heterozigot të një gjeni patologjik recesiv.
Metodat e ADN-së Rekombinante(Klonimi i ADN-së dhe hibridizimi i acidit nukleik)- ju lejon të zbuloni një gjen patologjik në gjenom nëse ky gjen është sekuencuar (sekuenca nukleotide është plotësisht e përcaktuar).
Analiza dermatoglifikeështë studimi i lëkurës së kurrizit të njeriut (lëkura e majave të gishtave, ana pëllëmbë e duarve dhe ana shputore e këmbëve), ku shtresa papilare e dermës është fort e theksuar.
Metoda aplikohet:
a) për të vendosur zigozitetin e binjakëve;
b) si metodë e shprehur për diagnostikimin e komponentit kongjenital të disa sëmundjeve trashëgimore.
Në mënyrë tipike, me patologji gjenomike, vërehet një kombinim i treguesve të caktuar: sythe radiale në gishtat e 4-të dhe të 5-të, zakoni katërshifror, këndi kryesor i palmës nga 60 0 në 80 0, etj.
Metodat kimike bazuar në reaksione kimike me ngjyra të cilësisë së lartë. Ato përdoren për diagnostikimin paraprak të sëmundjeve metabolike trashëgimore. Si një test depistues diagnoza e fenilketonurisë Përdoret metoda e njomjes së shiritave të letrës të njomur në një tretësirë 10% të FeCl 3 ose 2,4 dinitrofenilhidrazine me urinën e fëmijës. Nëse acidi fenilpiruvik është i pranishëm në urinë, në letrën filtri shfaqet një ngjyrë e gjelbër.
Përkufizimi i X- dheY-kromatina e seksit. Qelizat e epitelit bukal ose leukocitet përdoren për kërkime. X-kromatina përcaktohet duke ngjyrosur preparatin acetorcein, dhe Y-kromatin - kur ngjyroset Acrichinipritom. Këto metoda bëjnë të mundur identifikimin e numrit të kromozomeve seksuale në një kariotip (numri i kromozomeve X është gjithmonë një më shumë se numri i grupeve të kromatinës X, numri i kromozomeve Y është i barabartë me numrin e grupeve të kromatinës Y) ; përcaktoni seksin gjenetik të një individi, diagnostikoni sëmundjet kromozomale të seksit (në kombinim me metoda të tjera).
Metodat e diagnostikimit prenatal bëjnë të mundur identifikimin e defekteve trashëgimore të fetusit në fazat e hershme të shtatzënisë.
1. indirekte metodat (objekt i studimit është një grua shtatzënë).
2. Metodat e drejtpërdrejta ( objekt studimi është fetusi).
Metodat indirekte tregohen për të gjitha gratë shtatzëna dhe përfshijnë:
ekzaminim obstetrik dhe gjinekologjik;
ekzaminimi gjenetik mjekësor;
ekzaminimi serologjik;
studimi i përmbajtjes së proteinave specifike për embrion.
Niveli -FP është rritur me: kërcënim për abort; vdekja intrauterine e fetusit; papajtueshmëria me Rh midis nënës dhe fetusit; shtatzënia e shumëfishtë; hernia e kordonit të kërthizës; defekte të tubit nervor; teratoma sakrokoksigjeale. Nivel i ulët -FP vërejtur në sindromën Down.
Metodat e drejtpërdrejta të kërkimit: jo invazive (ultrasonografia) dheinvazive (amniocenteza, biopsia e vileve korionike, fetoskopia).
Tregohen metoda direkte jo-invazive për të gjitha gratë shtatzëna gratë (të sigurta për nënën dhe fetusin).
Ultrasonografi - përdorimi i ultrazërit për të marrë një imazh të fetusit dhe membranave të tij në një monitor. Metoda përdoret në javën 12-15 të shtatzënisë, kur formohen të gjitha sistemet e organeve të fetusit, dhe bën të mundur identifikimin e shtatzënive të shumëfishta dhe defekteve të mëdha (hidrocefalus, anencefali, aplazi të gjymtyrëve, etj.).
Janë kryer metoda invazive direkte sipas indikacioneve strikte (mosha e gruas është më shumë se 35 vjeç; rirregullime strukturore të kromozomeve tek njëri prej prindërve; fëmija i parë ka një sëmundje me një lloj trashëgimie recesive; shtatzënia për shkak të traumës psikologjike, sëmundjeve të rënda etj.).
Amniocenteza. Pas trajtimit të lëkurës, muri i barkut dhe mitra shpohen me gjilpërë dhe lëngu amniotik nxirret me shiringë nën kontrollin e ultrasonografisë. Lëngu përdoret për kërkime biokimike (diagnoza e mutacioneve të gjeneve), dhe qelizat fetale që përmbahen në të përdoren për kërkime citogjenetike (diagnoza e mutacioneve kromozomale dhe gjenomike).
Biopsia e vileve korionike kryhet nën kontrollin e ultrazërit në javën 8-12 të shtatzënisë. Duke përdorur një kateter me një kanulë plastike të lidhur me një shiringë, nxirren 10-25 mg villi korionike dhe përdoren për studime citogjenetike, biokimike dhe analiza të ADN-së. Metoda ju lejon të diagnostikoni mutacione të ndryshme në shtatzëninë e hershme.
Fetoskopi (amnioskopi) kryhet në javën 18-22 të shtatzënisë duke përdorur një endoskop fiberoptik të futur nën anestezi lokale në zgavrën e amnionit përmes murit abdominal të mitrës. Metoda lejon ekzaminoni fetusin, placentën, kordonin e kërthizës dhe merrni gjakun e fetusit nga kordoni i kërthizës për të diagnostikuar sëmundjet trashëgimore të gjakut. Përdoret rrallë; në 5–10% të rasteve mund të shkaktojë abort.







